

8月19日,由转化医学国家重大科技基础设施(上海)创新医疗器械注册研究与临床转化服务中心(NFTM-MRC)主办的上海交通大学长三角创新医疗器械注册研究与临床转化系列课程(YMRC)第12期在线上举行,出席论坛的有泰格捷通海外事业部杨怡斐总监、NFTM-MRC许苑晶等。
疫情的常态化、持续性蔓延对全球公共卫生系统、民众生产生活等带来了巨大影响,世界各地对口罩、防护服、呼吸机、检测试剂等医疗物资需求暴涨,这似乎给国内的相关企业带来出口海外的契机。但每个国家和地区的政策法规、监管要求不尽相同,市场准入和监管法规具有复杂性和多变性,把握合规性,才是决定出海可持续性的关键所在。在此背景下,杨怡斐总监带来了"医疗器械出海合规性的思考要素"的主题讲课。

杨怡斐总监从医疗器械出海的海外当地代理人/持证人、质量管理体系要求和注册/临床路径三个方面进行介绍。医疗器械的海外当地代理人/持证人由制造商审核和授权,作为当地监管部门与制造商之间沟通的窗口,海外当地代理人需协助制造商进行医疗器械的海外注册事宜、申请医保目录以及维护GMP证书等,并承担产品上市后不良事件监测、警戒报告的职责。海外当地代理人/持证人一般是国内医疗器械厂商在海外的分支子公司、海外经销商,或者当地专业的CRO公司等。

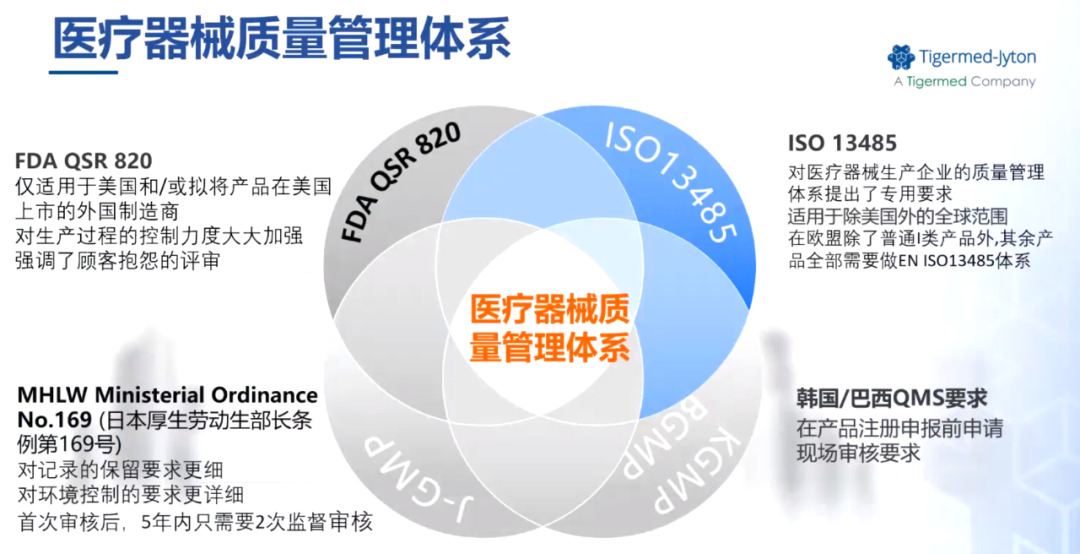
医疗器械的海外上市需按照各国的监管要求建立质量管理体系,并且保证其有效运行。质量管理体系覆盖了机构、人员、厂房设施、文件、设计开发、采购、质量控制、销售和售后服务、不合格品控制、不良事件监测和分析等要求,大体上符合ISO13485,但不同国家和地区的具体要求也存在差异。
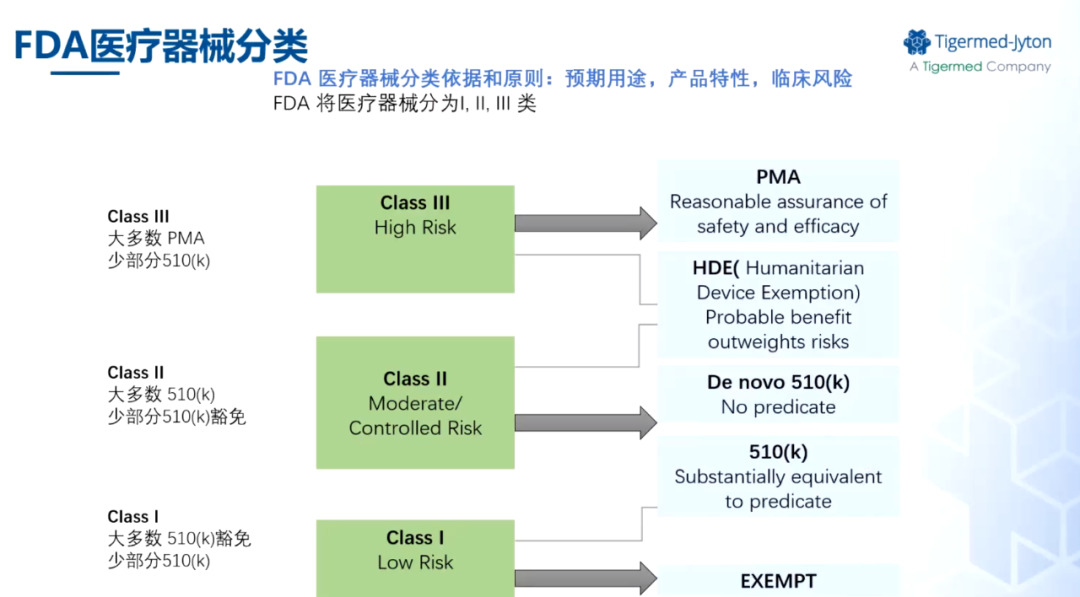
在注册方面,和国内一致,美国FDA将医疗器械分为三类,Ⅲ类风险管控等级高于Ⅰ、Ⅱ类。在510(k)递交过程中,申请者须证明新的器械与对比器械在预期用途、技术特征、性能测试方面实质等同,原则上不需要提交临床研究,大部分Ⅲ类器械要求的递交方式为PMA。De Novo针对无有效对比的新器械,HDE为用于罕见疾病治疗的器械提供了人道主义申请方法。

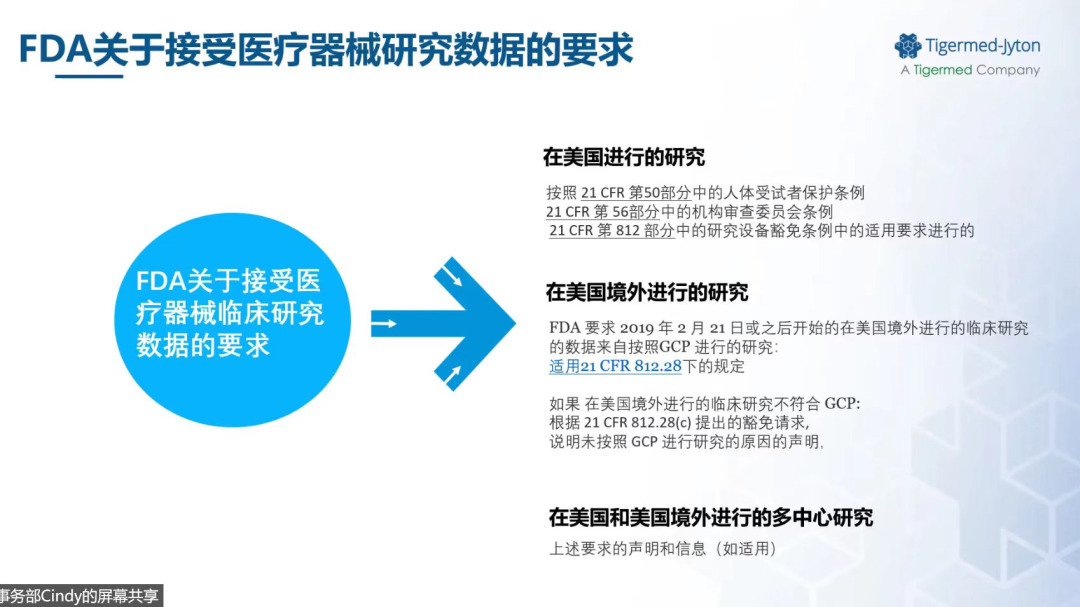
在临床评价方面,欧盟的医疗器械监管法规在过去的10~15年间发生了较大的变化,强调制造商需要基于临床数据和充分的临床证据来证实医疗器械可以达到预期的使用、评价非预期的副作用以及收益风险比例的可接受性等。临床评价还必须确认器械的预期用途及相关的临床收益、使用条件和特殊的禁忌症等,以上要求都需要临床证据的支持。临床评价也需要开展全生命周期的更新,而不是一次性就能完成的任务。


上海交大创新医械注册转化服务中心将持续提供优质的线上课程,所有线上学员均可申请成为上海交大转化注册中心会员,成为上海交大注册转化Family的成员,免费获得今后学术通知与生物医药产业转化服务的机会!


Copyright © 2022 Corporation All Rights Reserved
地址:上海市闵行区剑川路上海交通大学闵行校区 邮编:201203
沪交ICP备20220211 技术支持:戴院士王金武教授科研团队