

8月5日,由转化医学国家重大科技基础设施(上海)创新医疗器械注册研究与临床转化服务中心(NFTM-MRC)主办的上海交通大学长三角创新医疗器械注册研究与临床转化系列课程(YMRC)第10期在线上举行。出席课程的有NFTM-MRC副主任万克明, 斐卫(上海)医药科技有限公司临床资深培训师杨亦蕾等。课程聚焦医疗器械临床试验质量控制,主讲人杨亦蕾为临床药学专业背景,具有十年临床试验经验,熟悉医疗器械临床相关法规,现负责斐卫(上海)医疗器械临床试验项目管理及临床团队日常培训工作,擅长临床法规与实际临床案例结合。此次课程围绕临床试验研究数据和保障受试者权益两个方面,从方案偏离、临床试验器械、原始记录与报告和临床试验相关文件管理等进行了经验与方法分享。

主办单位NFTM-MRC由戴尅戎院士担任名誉主任,王金武教授担任中心主任,依托高校进行产学研医用相关标准、检测、注册研究与相关技术服务,并开展高校试制基地与监管科学的探索,旨在协助研团队加速医疗器械产业化。

医疗器械注册临床试验,是指在有资质的医疗器械机构中,对拟申请注册的医疗器械在正常使用条件下的安全性和有效性进行确认或者验证的过程。为深化医疗器械审评审批制度改革,加强医疗器械临床试验管理,国家药监局、国家卫生健康委在今年3月24日发布《医疗器械临床试验质量管理规范》(2022年第28号),并自5月1日起已正式施行。除了要遵循依法原则,临床试验还应当遵循《赫尔辛基宣言》提出的伦理原则和试验研究要求的科学原则。
根据ICH的定义, GCP是通过规定临床试验的设计、行为、性能、监测、审计、记录、分析和报告的标准,保证数据和报告的结果是可信和准确的,受试者的权利、完整性和机密性得到保护。临床试验的真实性是首要保障的,这不仅仅关系到器械的安全和有效是否经得起考验,对于参与的研究者来说,若临床试验被判定为含有虚造的“影子病人”等,除了需承担相关责任外,还可能被判定为学术不端。

“医疗器械临床试验是申办方实现产品上市的必经之路,在这条路上更要坚决保护受试者权益,才能体现产品本身的价值和临床试验的意义”,杨亦蕾总结道。知情同意书(ICF)是指向受试者告知医疗器械临床试验的各方面情况后,受试者确认自愿参加该项医疗器械临床试验,书面签署姓名和注明日期的证明文件。研究者需向受试者说明试验性质、目的、可能的受益和风险、权利和义务等,使受试者充分了解后表达其意愿。在临床试验的启动、执行、监查和关闭等环节,都应该注意ICF的记录和存档,若ICF在项目开展后进行更新升版,涉及到受试者权益改变等,应重新签署。
病历记录的质量决定了临床试验的质量,没有记录就没有发生。临床试验过程中产生的大量源数据,包括临床发现、观测结果以及用于重建和评价临床试验所需要的其他相关活动记录。源文件作为源数据的载体,包括了临床试验中产生的医院病历、医学图像、实验室记录、相关备忘录、受试者日记、评估表单、发放记录等,可以以纸质或者电子进行存档。

临床试验用器械管理常见问题包括:运输、接收、储存、分发、回收与处理记录不完整、签字不全,应填全管理记录表,在发放和回收记录处进行贴标等;无器械检验报告,当器械需要增加新批次入库,企业需要提供每个批次自检报告;温湿度记录不完整,特别是对环境敏感的器械,比如敷料产品,需要在规定的温湿度下进行保存以避免变质;对照产品文件不齐全等。
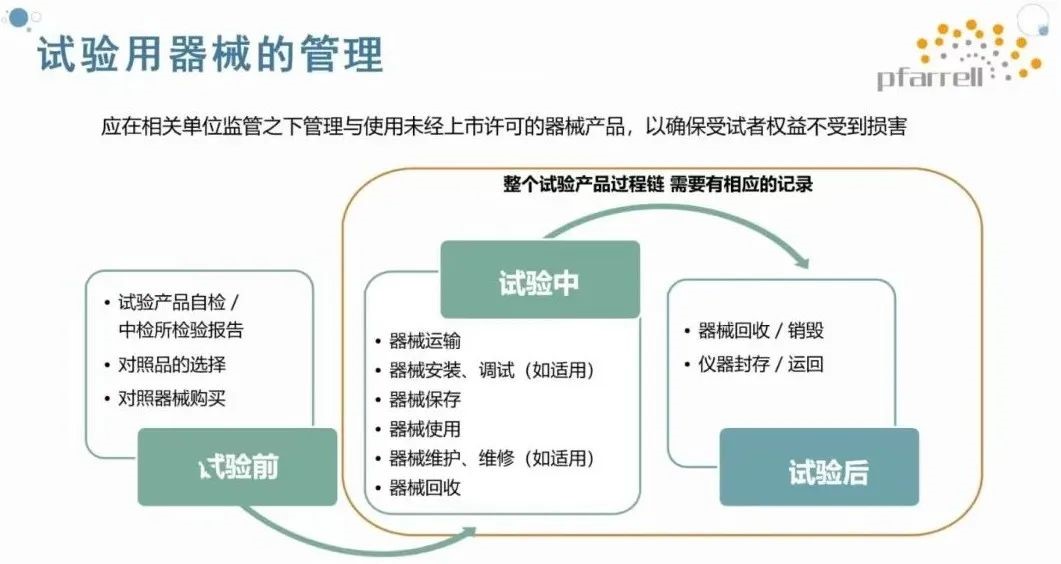
文件管理常见问题则包括:研究者资质不全,应搜集研究者GCP证书、简历等,确认执业地点在试验中心;培训记录不完整,当发生方案偏离后,应组织研究者培训并保留记录;试验物资校准证书过期;检查记录及报告缺失,临床试验输出的文件均应及时进行归档。

医疗器械临床试验质量控制应遵循医疗器械临床试验相关法规要求,制定完整的项目标准,确保过程实施中执行到位,申办者也需建立完善的质量体系。作为产品质量安全第一责任人,上市后企业也要持续关注临床使用风险。上海交大创新医械注册转化服务中心将持续提供优质的线上课程,所有线上学员均可申请成为上海交大转化注册中心会员,成为上海交大注册转化Family的成员,免费获得今后学术通知与生物医药产业转化服务的机会!


Copyright © 2022 Corporation All Rights Reserved
地址:上海市闵行区剑川路上海交通大学闵行校区 邮编:201203
沪交ICP备20220211 技术支持:戴院士王金武教授科研团队